Определение рассеянных элементов в морской воде методом нейтронного активационного анализа
Введение
Вода Мирового океана почти повсеместно отличается постоянством соотношений основных компонентов; в связи с этим изменение солености давно используется для идентификации водных масс. Однако изменение концентраций рассеянных элементов не зависит от вариаций солености, поскольку в процессах поступления и удаления участвует большая часть рассматриваемых элементов. Познание процессов геохимии моря, связанных с этими элементами, зависит от точности определений их малых количеств, присутствующих в морской воде.
Нейтронный активационный анализ является высокочувствительным методом, позволяющим определять большое число элементов. В последнее время этот метод использовался для выявления ряда элементов в морской воде (Fukai, Meinke, 1959). Настоящая работа была начата в связи с исследовательской программой, согласно которой намечалось определение возможно наибольшего числа рассеянных элементов в отдельном аликвотном объеме морской воды при определенных радиохимических условиях системы. Результатом этих работ явилась разработка более специализированных программ исследований, в которых основное внимание уделялось определению сравнительно ограниченного числа элементов при весьма широком отборе проб воды Мирового океана.
Методика отбора проб
Методика отбора проб частично была описана ранее в работе Болтера, Турекьяна и Шутца (Bolter, Turekian, Schutz, 1964). Пробы воды отбирались совместно с Ламонтской геологической обсерваторией во время двух рейсов экспедиционного судна "Вема" в 1961 и 1962 гг. Глубинные пробы воды отбирались преимущественно при помощи 50-галонного цилиндрического батометра с эпоксидным покрытием (Gerard, Ewing, 1961), по программе отбора проб для определения Cs137, Sr90, С14.
Как показано в табл. 5, некоторые пробы отбирались при помощи 50-галонного цилиндрического батометра без какого-либо специального покрытия, если не считать антикорродийной пленки металла, нанесенной гальваническим способом. Кроме того, ряд поверхностных проб был взят при помощи помпы, используемой для охлаждения гидролебедки. Подавляющая часть проб фильтровалась через микропористые фильтры с отверстием 0,45 μ, а аликвотная часть объемом 100 мл переносилась пипеткой в флаконы из стекла пирекс, снабженные притертыми пробками. Затем эти флаконы запечатывались электроизоляционной лентой и хранились до двух лет. По истечении определенного времени проводились контрольные опыты с использованием радиоактивного селена, серебра, кобальта, цезия, цинка, хрома и сурьмы. Было показано, что, по крайней мере, для этих элементов не наблюдается значительной адсорбции при периоде хранения до 6 месяцев, а для цинка, хрома и сурьмы - в течение 2 месяцев (табл. 1). Однако маловероятно, чтобы и при более длительных периодах хранения влияние адсорбции проявлялось столь значительно, что обесценило бы установленные величины аналитической ошибки. В определение аналитической ошибки входит и ошибка, обусловленная дифференциальной адсорбцией, поскольку такие оценки основаны в некоторых случаях на определении нескольких пар проб, время хранения которых различалось не менее чем на год.
| Элемент | Контрольная аликвотная часть, импульсы в минуту (7 марта 1963 г.) |
Испытуемая аликвотная часть, импульсы в минуту (21 сентября 1963 г.) |
% извлечения | Концентрация элемента в измеренной на счетчике пробе, мкг/л |
| Co | 664 | 667 | 100,5 | 0,5 |
| Ag | 1092 | 1078 | 98,6 | 0,5 |
| Cs | 13214 | 13027 | 98,5 | 1 |
| Se | 879 | 848 | 98,6 | 0,5 |
Пробы морской воды объемом 100 мл измерялись на регистрирующем устройстве и фильтровались через микропористый фильтр марки НА (0,45 μ) в склянки из стекла пирекс (125 мл). Параллельно с этим производился отбор контрольных аликвотных объемов (5 мл). На фильтрах не было обнаружено значительной активности.
Близкие результаты были получены для Zn и Cr после 2-месячного периода хранения и для Sb - после 3 недель.
Пробы из пролива Лонг-Айленд отбирались при помощи неметаллических пробоотборников, отфильтровывались через пористые фильтры с отверстиями 0,3 μ и хранились в тех же условиях, что пробы из глубоководных областей.
Активационный анализ
Требования к чувствительности метода
Концентрации подавляющего большинства рассеянных элементов в морской воде, как правило, меняются в интервале от мкг/л до 10-12 г/л. Для достижения необходимой аналитической чувствительности большинство исследователей бывает вынуждено прибегать к разнообразным методам химического концентрирования (Ishibashi et al., 1953; Lai, Weiss, 1962; Weiss, Lai, 1963; Weiss, Reed, 1960). В нашем исследовании не было необходимости в подобных подготовительных операциях, поскольку нейтронный активационный метод отличается чрезвычайно высокой чувствительностью. Таким образом, удалось избежать трудностей, связанных с операциями химического концентрирования.
Несмотря на то что некоторые рассеянные элементы можно определять при прямом облучении морской воды, чувствительность определений повышается в 30 раз при облучении выпаренного солевого остатка без какого-либо внесения химических реагентов. Однако выпаривание путем нагревания нередко осложняется разбрызгиванием, возможной летучестью некоторых рассеянных элементов и фракционной их кристаллизацией. Последняя, в частности, приводит к образованию неоднородной пробы. Все эти трудности удалось устранить при высушивании проб морской воды путем ее вымораживания. Этот метод кратко описан в работе Болтера, Турекьяна и Шутца (Bolter, Turekian, Schutz, 1964).
Проба морской воды объемом 100 мл быстро замораживалась в виде тонкого слоя на стенках вакуумной склянки (500 мл) при ее непрерывном вращении в бане из сухого льда и ацетона. Затем вода испарялась в вакуумной системе, снабженной ловушкой из сухого льда - этил-бутил-целлозольв. Полученный сухой весьма тонкозернистый порошок упаковывался тем или иным способом в зависимости от требований, обусловленных конструктивными особенностями облучающей установки. В нашем исследовании пробы помещали в алюминиевые ампулы диаметром 0,5 дюйма; герметическое закупоривание их производилось при помощи гидравлического пресса. Большинство образцов облучалось в контейнере на 60-70 проб в реакторе мощностью 5 Мвт, принадлежащем компании "Юнион кэрбид нуклеар" (Таксидо, Нью Йорк); реактор был установлен в плавательном бассейне. Несколько проб морской воды, заключенных в емкостях из алюминиевой фольги, облучалось в реакторе с воздушным охлаждением (Брукхейвенская национальная лаборатория).
Ограничения в определении долгоживущих продуктов активации
Чувствительность частично является функцией от времени облучения и периода полураспада полученных радиоактивных изотопов. Значительные количества натрия в составе солей морской воды и относительно малый период полураспада Na24 (15 часов) обусловливают либо весьма короткий период облучения, либо длительное время облучения, сопровождающееся продолжительным периодом охлаждения. Эти меры позволяют относительно легко оперировать пробами во время анализа. Конструктивные особенности реактора не позволяют получать короткоживущие изотопы; поэтому для получения результатов высокой чувствительности пробы подвергались длительному облучению (150 часов) при высокой интенсивности облучающего потока (2-6 X 1013 nV). Образующиеся при такой обработке большие количества Na24 обусловили необходимость продолжительного периода охлаждения (более 10 дней), что, таким образом, позволило изучить только относительно долгоживущие изотопы, указанные в табл. 2. Другие долгоживущие элементы, например металлы из группы платины и редкие земли, выделены из настоящего исследования и рассматриваются отдельно.
Радиохимическое разделение
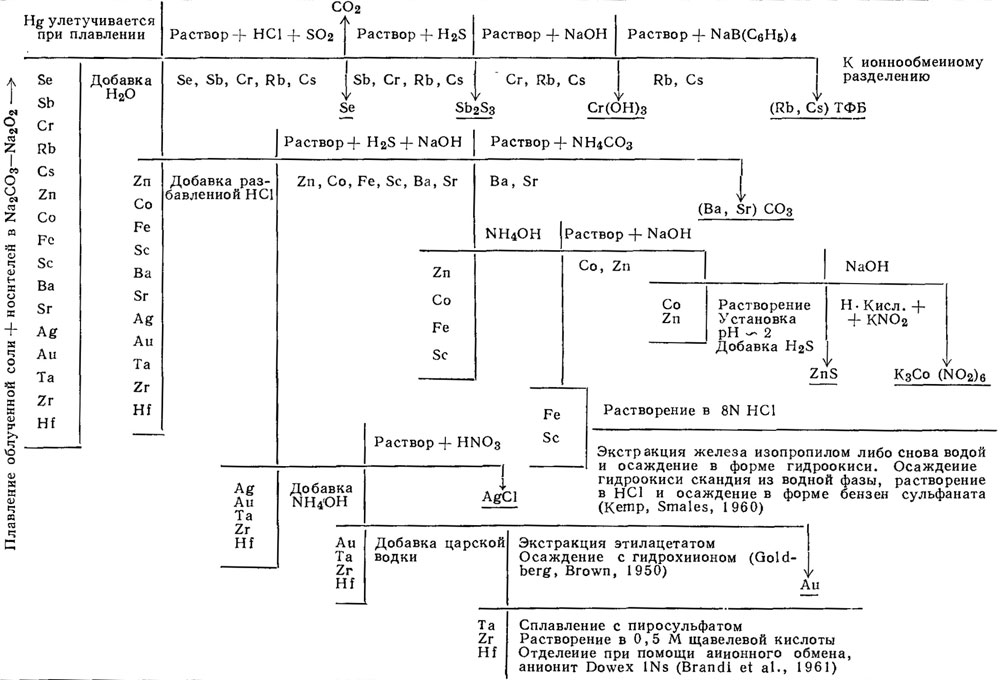
Рис. 1. Схема последовательности операций в анализе, основанном на методе равновесного плавления.
Дальнейшее ограничение в схеме анализа было связано с наличием хлора в составе солей морской воды. При продолжительном облучении, которое необходимо для получения высокой чувствительности, в результате термической нейтронной реакции Сl35 (n, р) S35 образовалось примерно 0,1-0,5 см3 S35 (период полураспада 87 дней, β-). Значительная величина активности тормозного излучения в облученной пробе и очень низкие активности интересующих нас изотопов привели к необходимости использования носителей и радиохимических методов сепарации.
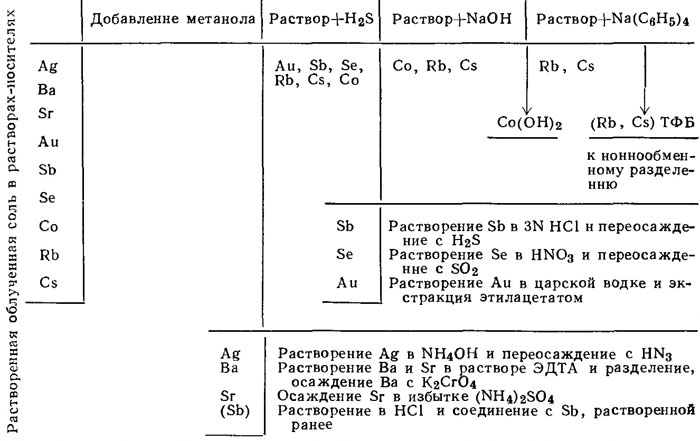
Рис. 2. Схема последовательности операций в анализе, основанном на методе равновесного растворения.
На рис. 1 и 2 показаны две разработанные нами схемы последовательности операций радиохимического разделения. Схема, основанная на плавлении Na2CO3 - Na2O2 (рис. 1), является более общей и может быть использована, помимо морской воды, для многих других минеральных соединений. Другая схема, начинающаяся с установления равновесия раствора (рис. 2), была создана для более быстрого определения сравнительно небольшого числа элементов.
| Реакция | Период полураспада |
Энергия γ-лучей, Мэв |
Экспериментальный предел*, определения, мкг/л |
| Ag109 (n, γ)Ag110 | 249 дн. | 0,66;0,88 | 0,02 |
| Au197 (n, γ)Au198 | 64,8 часа | 0,412 | 0,0001 |
| Ba130 (n, γ)Ba131 | 11,6 дн. | 0,63 | 2,5 |
| Co59 (n, γ)Co60 | 5,2 года | 1,17;1,33 | 0,005 |
| Cr50 (n, γ)Cr51 | 27,8 дн. | 0,32 | 0,01 |
| Cs133 (n, γ)Cs134 | 2,1 года | 0,60 | 0,05 |
| Fe58 (n, γ)Fe59 | 45 дн. | 1,10;1,29 | 1 |
| Hf174 (n, γ)Hf175 | 70 дн. | 0,343 | 0,005 |
| Hf180 (n, γ)Hf181 | 42,5 дн. | 0,482 | * |
| Hg202 (n, γ)Hg203 | 47 дн. | 0,279 | 0,05 |
| Ni58 (n, γ)Co58 | 71 дн. | 0,82 | 0,1 |
| Rb85 (n, γ)Rb86 | 18,7 дня | 1,08 | 0,2 |
| Sb123 (n, γ)Sb124 | 60 дн. | 0,60 | 0,05 |
| Sc45 (n, γ)Sc46 | 84 дня | 1,12 | 0,004 |
| Se74 (n, γ)Se75 | 120 дн. | 0,40 | 0,04 |
| Sr84 (n, γ)Sr85 | 64 дня | 0,51 | 24 |
| Ta181 (n, γ)Ta182 | 115 дн. | 1,12;1,22 | 0,0025 |
| U235 (n, γ)Ba140 | 12,8 дня | 0,81 (La140) | 0,1 |
| Zn64 (n, γ)Zn65 | 245 дн. | 1,14 | 0,3 |
| Zr94 (n, γ)Zr95 | 65 дн. | 0,72; 0,76 | 0,1 |
* (Весьма приблизительные пределы установлены на основании эксперементов, протекавших в типичных условиях: а) для определения необходимо 25 импульсов в минуту; б) поток=2Х1013 nV при продолжительности облучения 150 час; в) 100 мл пробы с 30%-ным химическим выходом; г) 14 дней охлаждения после выемки проб из реактора.)
Химические методы, применяемые в схемах разделения, являются либо стандартными, либо несколько модифицированными в сравнении с заимствованными из литературы методиками. В связи с этим мы остановимся лишь на вопросах, касающихся системы морской воды, окончательной очистки и последних этапов в определении отдельных элементов. Полная аналитическая методика описана в работе Шутца (Schutz, 1964). В целом наличие большого числа элементов в массе носителя (около 20-30 мг каждого металла) зачастую позволяет обходиться без "обратных" носителей. Более общей проблемой является определение S35. Метод, основанный на сравнении со стандартами, рассматривается ниже.
Ртуть. Улетучивающаяся при плавлении ртуть конденсируется в трубчатом отростке воронки, укрепленной в перевернутом виде над тиглем. Затем ртуть вымывается из воронки концентрированной HNO3, осаждается в форме сульфида, очищается путем ее кипячения с HNO3 (1:1), отфильтровывается на бумажный диск, высушивается, взвешивается и помещается в никелевую пластиновидную емкость для подсчета числа импульсов.
Селен. Селеновый осадок окклюдирует большие количества S35 в форме сульфата, который нельзя устранить даже при повторном осаждении в присутствии сульфатзадерживающего "обратного" носителя. Полная очистка от примесей достигается при помещении селенового осадка в кварцевую трубку, один конец которой запаян. При нагревании этой трубки селен отгоняется во взвешенную счетную ампулу из стекла пирекс. При взаимодействии с HNO3 селен окисляется в SeO2, высушивается и взвешивается. Затем SeO2 растворяется в воде и доводится до стандартного объема для подсчета импульсов цилиндрическим детектором.
Сурьма. Осадок Sb2S3 растворяется в НСl. Затем сульфид переосаждается для полного очищения от S35, после чего сурьма осаждается с 8-гидроксихинолином (Maeck, 1961); после высушивания при 110°, охлаждения и взвешивания осадок переносится в счетную ампулу, растворяется в 6N НСl и доводится до стандартного объема для подсчета.
Хром. Осадок Cr(ОН)3 захватывает большие количества S35, которую можно удалить лишь путем окисления хрома в бихромат при помощи Н2O2 в сильнощелочной среде и при последующей экстракции хромата метилизобутилкетоном (4-метил, 2-пентанон) из 3М НСl (Maeck et al., 1962). Следует провести полное разложение Н2O2, прежде чем подкислять раствор, во избежание восстановления хроматного иона. Последовательная очистка органической фазы аликвотными объемами 3М НСl позволяет полностью освободиться от примесей S35. Наконец, хромат вновь экстрагируется 2М NaOH - 3М NH4OH и осаждается в форме хромата бария. Отсутствие активности S35 убедительно свидетельствует о полном удалении сульфата из осадка ВаСrO4. Осадок высушивается, взвешивается, переносится в ампулу для подсчета, растворяется и доводится до стандартного объема.
Рубидий и цезий. Операции по определению этих двух элементов методом равновесного растворения подробно описаны в работе Болтера, Турекьяна и Шутца (Bolter, Turekian, Schutz, 1964). Осадок (Rb, Cs) тетрафенилбора нагревается в течение 30 минут при 350° и растворимые бораты отделяются с водой. Рубидий и цезий отделяются от калия и совместно осаждаются из морской воды при добавлении Na2AgBi (NO2)6. Чтобы добиться хорошего разделения, осаждение повторяют. Осадок (Rb, Cs) AgBi (NO2)6 растворяют в концентрированной HNO3, а висмут и серебро отделяют с H2S. Рубидий и цезий разделяют на колонке с катионитной смолой марки Dowex 50 W-xPs (200-400 мешей), используя в качестве растворителя для элюирования 0,6N HCl (Ishibashi et al., 1959). Затем рубидий и цезий осаждаются с натриевым тетрафенилбором, высушиваются и помещаются в никелевую пластиновидную емкость для подсчета импульсов.
Серебро. Хлорид серебра растворяется в NH4OH и затем очищается путем соосаждения с Fe(OH)3, что позволяет полностью избавиться от примесей S35. AgCl переосаждается при добавке HNO3. Осадок переносится на взвешенный микропористый фильтр, высушивается, взвешивается и растворяется для дальнейшего подсчета.
Золото. Осадок золота промывается на микропористом диске фильтра, прокаливается, взвешивается, переносится в мерную складку и растворяется для подсчета. Оставшуюся после начальной экстракции золота жидкую фазу подщелачивания, для того чтобы уловить в ней тантал, цирконий или гафний, которые могли раствориться при экстракции царской водкой.
Цирконий, гафний и тантал. Щавелевокислый раствор циркония, гафния и тантала пропускается через крошку с ионнообменной смолой типа Dowex 1-S (100-200 мешей по методу, описанному в работе Брэнди и др. (Brandi, et al., 1901). Затем цирконий и гафний совместно элюируется 200 мл раствора 1,5М НСl, 0,5М щавелевой кислоты и 0,007М Н2O2. Дополнительные 500 мл того же раствора элюируют Nb95 - продукт распада Zr95; этот раствор за ненадобностью выливается. Тантал элюируется 350 мл раствора 3М НСl и 0,5 М щавелевой кислоты. Элюант, содержащий цирконий и гафний, доводят до щелочной реакции. Осаждающиеся гидроокиси растворяют в НСl. Соответствующие соединения миндальной кислоты (тетраманделаты) осаждаются (Steinberg, 1960), прокаливаются до смешанных окислов, которые затем взвешиваются и измеряются совместно (Merz, 1962). Опыты с использованием меченых атомов показывают, что отношение Zr/Hf в конечном продукте не отличается от подобного отношения в смеси носителя, что делает возможным точное определение выхода. Это обстоятельство позволяет выполнять точные определения. Осадок циркония следует измерять сразу же после ионнообменной очистки, чтобы избежать побочного влияния нарождающегося Nb95. Тантал осаждается непосредственно из щавелевокислого элюанта при добавлении дубильной кислоты (Hillebrand et al., 1953, p. 604) после прибавления насыщенного раствора хлорида аммония. Отделенный таким способом тантал не содержит заметной примеси, что позволяет прокаливать осадок до окиси Та2O5 и взвешивать. После взвешивания осадок полностью переносился в пластиковую мерную трубку диаметром 16 мм, растворялся в HF и доводился до стандартного объема.
Цинк. Осадок сульфида цинка растворяют, переосаждают и прокаливают до окиси. Полученный прокаленный осадок взвешивается, переносится в мерную капсулу и растворяется в НСl.
Кобальт. Co2S3 растворяется в HNO3, осаждается в форме K3Co(NO2)3 и переосаждается вновь для отделения всей имеющейся S35. Осадок затем высушивается при 110° (Bate, Leddicotte, 1961), переносится с фильтра, взвешивается, помещается в мерную капсулу и растворяется в HNO3.
Железо. Каждая фаза, содержащая железо, тщательно очищается при помощи 8N НСl для полного устранения примесей S35. Затем железо снова экстрагируется в воду; гидроокись осаждается, прокаливается до окиси, которая охлаждается, взвешивается и растворяется для измерения.
Скандий. Неорганическая фаза, содержащая скандий, подщелачивается аммиаком. Затем гидроокись скандия растворяется в 3N НСl и осаждается при добавлении 4 мл 4%-ного раствора бензолсульфокислого натрия (Kemp, Smales, 1960).
Барий. Подготовительные операции метода равновесного растворения подробно описаны Болтером, Турекьяном и Шутцем (Bolter, Turekian, Schutz, 1964). BaCrO4 растворяется в уксусной кислоте и переосаждается в присутствии стронциевого тормозящего носителя и ЭДТА по методу Монтгомери (Montgomery, 1960). Этим способом было достигнуто прекрасное отделение от относительно высокой и частично совпадающей активности S35.
Стронций. Установлено, что для получения конечного осадка сульфата стронция, свободного от природного кальция морской воды, весьма эффективны два метода. Согласно первому методу, осадок карбоната стронция переводится в нитрат и кальций удаляется при смешении в пропорции 1:1 либо с этанолом, либо с ацетоном (Leliaert, Eeckhaut, 1957). Подобная обработка использовалась Болтером, Турекьяном и Шутцем (Bolter, Turekian, Schutz, 1964). В другом методе производится осаждение стронция в присутствии ЭДТА. Последний связывает кальций в более прочные комплексы, чем стронций (Montgomery, 1960). Избыток сульфатного иона, требующегося согласно этому методу, также служит, как "обратный" реагент в отношении S35O2+4, который может проходить через всю схему аналитического определения.
Опыты с использованием меченых атомов
Достижение равновесия и методика определения выхода контролировались экспериментами с мечеными атомами, результаты которых приводятся в табл. 3.
| Элемент | Метод плавления | Метод растворения | ||
| число проб | хим./радиохим. средний выход |
число проб | хим./радиохим. средний выход |
|
| Ag | 3 | 1,02 | 3 | 1,01 |
| Au | 4 | 0,99 | 2 | 1,01 |
| Ba (U-Ba140) | 4 | 0,98 | 2 | 0,99 |
| Co (Ni-Co58) | 4 | 1,00 | 3 | 0,99 |
| Cr | 2 | 1,01 | - | - |
| Cs | - | - | 3 | 0,975 |
| Fe | 3 | 0,99 | - | - |
| Hf-Zr | 2 | 1,06 | - | - |
| Hg | 3 | 1,01 | - | - |
| Rb | - | - | 3 | 0,975 |
| Sb | 3 | 1,02 | 3 | 0,97 |
| Sc | 2 | 1,03 | - | - |
| Se | 2 | 0,98 | 3 | 0,99 |
| Sr | 3 | 1,02 | 3 | 1,01 |
| Ta | 2 | 1,04 | - | - |
| Zn | 3 | 1,03 | - | - |
Вещество основной массы, использовавшееся в опытах с мечеными атомами, отличалось от вещества облученной соли лишь наличием S35. Возможные побочные загрязнения проб при опытах с мечеными атомами и проб морской воды постоянно контролировались параллельными разделениями неактивного материала. Ни в одном из этих холостых опытов не было обнаружено ощутимой активности, даже при работе с мечеными атомами, активность которых нередко значительно превышала активности в необлученных пробах морской воды.
Методика измерения
Очищенная форма каждого элемента замерялась при помощи обычного 1,5X2 дюйма или цилиндрического 3Х3 дюйма, NaI (Tl) сцинтилляционного счетчика совместно с амплитудным анализатором импульсов модели 402 производства "Текникал межемент корпорейшн" (ТМК).
Комплексные спектры, в частности суммарные спектры Со58 и Со60, используемые соответственно при определении никеля и кобальта, а также суммарные спектры Ва140 и Ва131, используемые при определении урана и бария, разрешались при помощи интегрирующего устройства модели 522 производства ТМК. В этом устройстве имелись также соответствующие приспособления для снятия фона и единичного сложения площадей γ-пиков. Окончательные спектры γ-лучей проверялись на чистоту путем сравнения со стандартными источниками, а в случае более короткоживущих изотопов путем определения периода их полураспада.
Регистраторы потока
Проведению активационного анализа мешает не только хлор морской воды, из которого образуется S35 и который по этим причинам необходимо отделять методом радиохимического разделения. На результаты активационного анализа существенно влияет также и самоэкранирующий эффект каждого образца из-за большого сечения активации Сl35. Подсчитано, что этот ослабляющий эффект составляет 7%. Эта цифра согласуется с результатами измерения четырех проб, в которые были внесены известные количества серебра и кобальта; затем было проведено сравнение с внешним регистратором потока. Величины для первых образцов, анализированных при помощи внешних регистраторов потока, подсчитывались с учетом такого коэффициента самоэкранирования, который связан с внесением поправок на количество использованной воды, ее исходной солености и потери соли во время испарения и упаковки. Однако постоянная величина отношения стронция к солености для широкого интервала соленостей и различных географических точек (Chow, Thompson, 1955) позволяет использовать стронций как внутренний регистратор потока, не прибегая к этим поправкам (Bolter, Turekian, Schutz, 1964). Этот метод основан на допущении, что отношение удельной активности стронция и данного элемента в пробе равно отношению удельных активностей этих элементов в стандартах, совместно облученных снаружи. Проверялась также возможность изменения этого отношения вследствие резонансной адсорбции эпитермических нейтронов. Проверка проводилась путем сравнения стандартных пар отношения стронция к элементу, в которых опытные интенсивности потока различались вдвое. Наибольшие различия (7%) были установлены для реакции Ni58 (n, р) Со58, чего и следовало ожидать из-за относительно высокого значения порога этой реакции. Аналогичны и все остальные отношения в пределах статистически подсчитываемых величин для обоих местонахождений. Все стандартные регистраторы потока готовились путем перенесения пипетками спектрально чистых растворов фирмы "Джонсон - Мэттхэй" в чашки из алюминиевой фольги, отличающиеся высокой чистотой.
Другие применения метода
Несмотря на то, что первоначальной целью рассматриваемого метода являлся анализ морской воды, способ равновесного сплавления позволяет широко применять этот метод для анализа малых количеств практически любого материала, в котором предстоит определение рассеянных элементов. Этим методом можно анализировать космическую пыль, поровые воды глубоководных осадков и многое другое.
Метод внутренних стандартов применим и к другим системам, для которых имеются подходящие природные стандарты либо к которым можно добавить изотопы, не нарушающие этой системы. Однако подобные меры могут оказаться необходимыми лишь в тех случаях, когда появляются трудности, связанные с сильными эффектами самоэкранирования и неоднородностями потока.
|
ПОИСК:
|
При использовании материалов сайта активная ссылка обязательна:
http://iznedr.ru/ 'Из недр Земли'